全国咨询热线:
0755-23699400

器械与放射健康中心(CDRH)负责美国医疗器械的管理。根据医疗器械风险将产品分为I,II,III类。在美国的上市途径主要包括:
1) Premarket Notification (510(k))(即:上市前通告),
2) De Novo,
3) Exempt(豁免上市前通告)
4) Premarket Approval (PMA)(即:上市前批准),
5) Humanitarian Device Exemption (HDE)(即:人道主义器械豁免)。
其中大部分I类产品的上市途径为510KExempt豁免上市前通告即直接注册列名;
大部分II类和部分III类产品上市途径为上市前通告PMN,即510(K);
部分IIII类产品上市途径为上市前批准PMA。通过PMA获得上市批准的产品,需通过美国FDA按照21CFR 820要求的质量体系审查。
针对上市途径为510(K),但无等同器械,需向FDA提交De novo获得上市前通告。
美国FDA的证书无有效期,获得上市许可的企业需在美国FDA官网进行企业注册和产品列名。
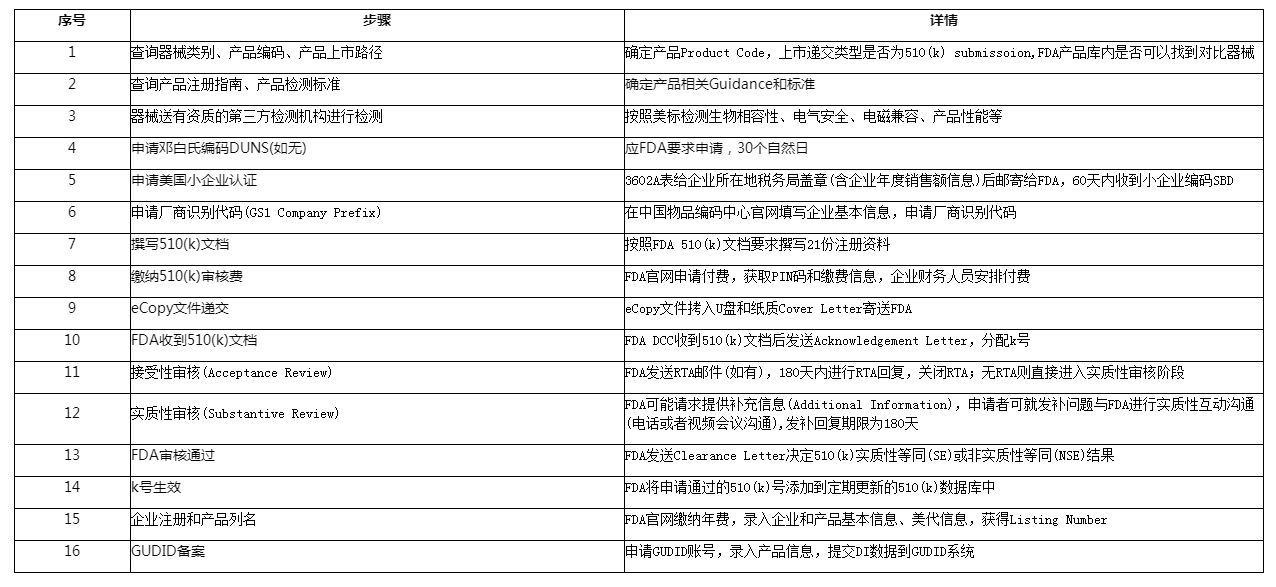
流程图:
以510(k)注册为例
通用法规:
美国法规和指南
官方时间和费用:
官方时费用(单位:万元)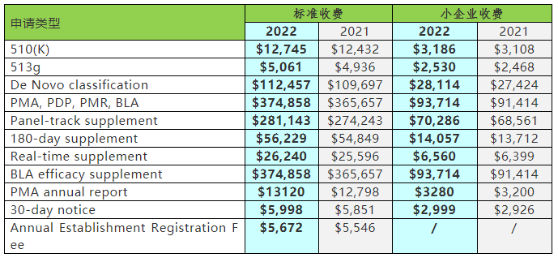
官方时间
510(K)审批时间:180天
De novo审批时间:120天
Pre-Submission审批时间:30天
PMA审批时间:225天
相关业务:
上市前批准(PMA)
创新器械上市(De Novo)
上市前通告510(K)Submission
企业注册和产品列名
美国代理人服务
QSR820验厂辅导
服务热线

 全国服务热线
全国服务热线