
欧盟将医疗产品分为医疗器械MD及体外诊断医疗器械IVD。
医疗器械条例(MDR EU 2017/745) 根据器械的预期用途和固有风险将产品分为I类(Is类),IIa, IIb,III类。其中I类采用自我符合性评估获得上市许可,其他类别需通过NB公告机构进行符合性评估获得上市许可,获得上市许可前需通过公告机构按照ISO 13485标准的质量体系认证。CE证书有效期为5年。
体外诊断医疗器械(IVDR 2017/746)根据风险等级将产品分为A(A类无菌),B,C, D四类。其中A类采用自我符合性评估上市,其他类别需通过公告机构进行符合性评估获得上市许可。获得上市许可前需通过公告机构按照ISO 13485标准的质量体系认证。CE证书有效期为5年。
流程图:
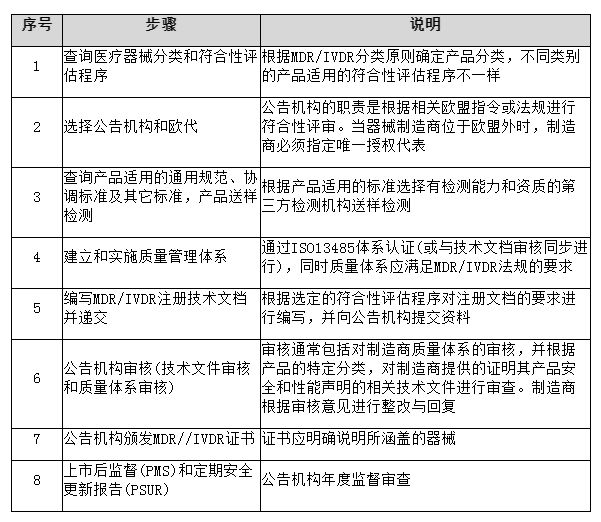
通用法规:
EU 2017/745法规 ( MDR)
EU 2017/746法规 (IVDR)
官方时间和费用:
官方费用
以公告机构的价格为准;
官方时间
MDR
I 类审批:1个月
Is类,IIa和IIb审批:6个月
III类审批:8-10月
IVDR
A 类审批:1个月
B类和C类审批:6个月
D类审批:8-10月
相关业务:
MDR&IVDR技术文件编写
欧盟临床评价
欧盟代表服务
ISO 13485质量体系辅导
